О направлении
Проводимая нами работа была направлена на изучение внутриклеточных сигнальных систем, которые обуславливают цитотоксическое действие цитокинов на бета-клетки или, напротив повышают устойчивость клеток к патогенным факторам. Фармакологическим подходом для защиты бета-клеток может быть избирательное воздействие на зависимые от цитокинов сигнальные системы и их ключевые элементы, такие как транскрипционные факторы HIF-1α, AP-1 и Nrf2, протеинкиназы AMPK и c-Abl, фактор трансляции eIF5A.
Также мы исследовали роль адаптерного белка β-аррестин-1 в контроле внутриклеточной локализации рецептора межклеточного сигнального фактора GLP1 в бета-клетках. Известно, что GLP1 повышает секрецию инсулина и устойчивость бета-клеток к патогенным воздействиям. При связывании с лигандом происходит обратимая инактивация мембранного рецептора GLP1 и β-аррестин-1 участвует в этом процессе.
Более ранние темы (до 2011).
Были исследованы сигнальные системы, контролирующие экспрессию генов, чувствительных к АФК, АФА и ксенобиотикам, в частности MAP киназы, тирозиновые киназы семейства Src, факторы транскрипции Nrf2, NF-κB и AP-1.
В итоге был определен спектр и обнаружены новые зависимые от NO гены в культуре первичных хондроцитов и клетках моноцитарного происхождения. Изучено влияние гипоксии, а также антиоксидантов и других соединений, влияющих на образование АФК и АФА и индукцию зависимых от NO генов. Значительная часть зависимых от NO генов меняет свою экспрессию также при гипоксии, окислительном стрессе и под действием электрофильных ксенобиотиков. Вместе с тем, в целом выявленный нами спектр зависимых от NO генов уникален и не совпадает со спектрами генов, индукция которых происходит при других воздействиях на клетки. Зависимые от NO гены можно отнести к разным функциональным группам. Они участвуют в регуляции апоптоза, клеточного роста, синтеза белка и транскрипции. Зависимые от NO гены различаются по пороговым концентрациям NO, при которых происходит их индукция.
Наши исследования показали, что в моноцитах транскрипционный ответ клеток на NO опосредован промежуточной генерацией АФА и модификацией рецепторных SH-групп. Выявлены сигнальные системы, участвующие в транскрипционной активации зависимых от NO генов в клетках моноцитарного происхождения. Исследованы соединения (например, ароматические метиленмалононитрилы, АМН), способные избирательно модифицировать тиольные группы рецепторных белков, стимулировать регуляторную систему Keap1/Nrf2, подавлять экспрессию про-воспалительных генов и повышать устойчивость клеток к цитотоксическим воздействиям. Эти соединения отчасти воспроизводят клеточный ответ на NO.
В культурах панкреатических бета-клетках наряду с индукцией защитных генов (например, гемоксигеназы 1, HO-1) соединения АМН подавляют зависимую от про-воспалительных цитокинов (IL-1β и IFNγ) активацию экспрессии гена синтазы окиси азота (iNOS). Наряду с соединениями АМН ответ бета-клеток на цитокины полностью блокирует брусатол. В присутствии брусатола происходит подавление ряда ответных реакций бета-клеток на действие про-воспалительных цитокинов: апоптоза, окислительного стресса, активации экспрессии iNOS и нарушения секреции инсулина. Брусатол ингибирует экспрессию iNOS на посттранскрипционном уровне (предположительно, через подавление гипузинирования трансляционного фактора eIF5A).
Ранее нами были исследованы контролируемые интерферонами внутриклеточные системы. В частности, регуляция экспрессии и активности 2-5 олиго(А)синтетазы и триптофанил-тРНК-синтетазы (сокр: WRS) и метаболизм образуемых этими системами уникальных 2’,5’ и 5’5’ олигоаденилатов. Кроме того, изучался альтернативный сплайсинг про-мРНК гена WRS. Также проводилась работа по поиску и описанию ДНК-связывающих белков специфичных к регуляторным участкам генов.
Методы исследования.
Проточная цитометрия, блот-гибридизация белков, количественная ПЦР (RT-PCR), иммунопреципитация белков, ELISA, трансфекция клеточных культур и интерференция РНК. Также: хроматография олигонуклеотидов и белков (TLC, HPLC), очистка и фракционирование белков, молекулярное клонирование, секвенирование ДНК, исследование ДНК-связывающих белков методами сдвига электрофоретической подвижности (band-shift assay) и футпринта (protein/DNA footprint), масс-спектральный анализ.
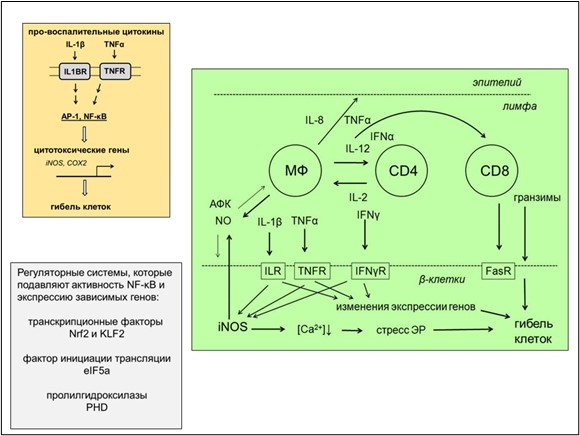
Рисунок 1. Сигнальные системы участвующие в развитии аутоиммунного диабета. Левая верхняя панель. Обобщенная схема ответа бета-клеток на про-воспалительные цитокины. Цитотоксическое действие про-воспалительных цитокинов опосредовано активацией факторов транскрипции AP-1 и NF-κB и индукцией контролируемых ими генов (синтаза окиси азота iNOS, циклооксигеназа-2 COX2 и др.). Сокращения: IL1BR и TNFR – рецепторы цитокинов IL-1β и TNFα, соответственно. Правая верхняя панель. Схема цитотоксических процессов при диабете 1 типа. После первичной активации, в которой участвуют дендритные клетки (не показаны), цитотоксические лимфоциты через эндотелий кровеносных сосудов проникают в заполненное лимфой пространство островков поджелудочной железы. Там лимфоциты вступают в сложное взаимодействие между собой и секретирующими инсулин бета-клетками. В сигнальных процессах участвуют цитокины, хемокины (IL-8), АФК и АФА. Во многом эти процессы координируются по принципу положительной обратной связи. Про-воспалительные цитокины и окислительный стресс усиливают миграцию и созревание лимфоцитов, стимулируют дальнейшую генерацию цитокинов, хемокинов, АФА и АФК, а также инициируют адгезию между цитотоксическими Т-лимфоцитами CD8+ и бета-клетками. При действии про-воспалительных цитокинов на макрофаги (МФ) и бета-клетки в них происходит индукция iNOS и рост выработки NO. Действуя на лимфоциты NO усиливает секрецию ими про-воспалительных цитокинов, действуя на бета-клетки – способствует снижению уровня ионов Ca2+ в эндоплазматическом ретикулуме (ЭР), что вызывает стресс ЭР и индуцирует апоптоз. Гибель бета-клеток также обусловлена зависимыми от цитокинов изменениями экспрессии генов, прежде всего теми, которые контролирует фактор NF-κB. Большое значение также имеет активация цитотоксических Т-лимфоцитов CD8+ и секреция ими токсических гранзимов. При прямом контакте Т-лимфоцитов CD8+ и бета-клеток гибель последних может быть обусловлена активацией рецепторов Fas (FasR). ILR, TNFR, IFNγR – рецепторы цитокинов IL-1β, TNFα и IFNγ, соответственно. На левой нижней панели перечислены регуляторные системы, способные предотвращать гибель β-клеток под действием про-воспалительных цитокинов.
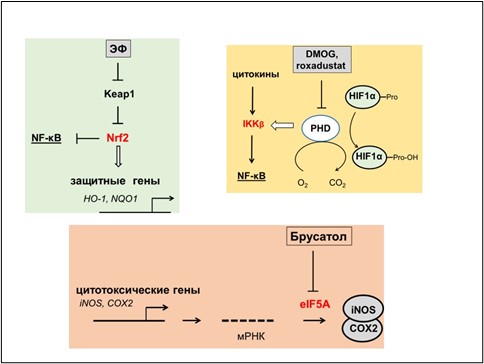
Рисунок 2. Фармакологические подходы для предотвращения аутоиммунного диабета. Ослабление в β-клетках токсических эффектов цитокинов при подавлении сигнальных систем и индукции генов, зависимых от IL-1β и TNFα. Электрофильные соединения (ЭФ) активируют транскрипционный фактор Nrf2, что снижает связывание NF-κB с ДНК и экспрессию зависимых генов. Кроме того, Nrf2 индуцирует гены, повышающие устойчивость клеток к окислительному стрессу (HO-1 – гемоксигеназа-1, NQO1 - NADH-хиноноксидлредуктпзп-1 и др.). Также подавляет активность NF-κB пролилгидроксилаза PHD. Основная функция PHD как сенсора кислорода заключается в негативной регуляции экспрессии чувствительных к гипоксии генов, что обусловлено подавлением транскрипционного фактора HIF-1α в условиях нормоксии. DMOG и roxadustat – фармакологические активаторы PHD при нормоксии. На уровне синтеза белка для экспрессии про-воспалительных генов необходим фактор трансляции eIF5A. Брусатол (растительный алкалоид) ингибирует eIF5A и блокирует трансляцию мРНК гена iNOS.
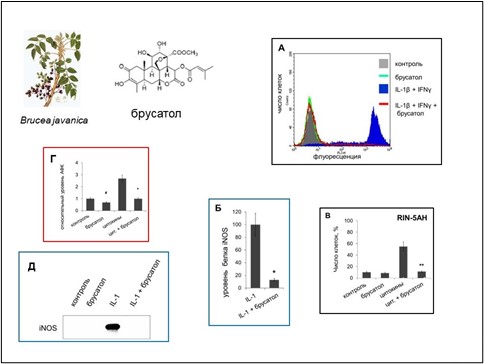
Рисунок 3. Брусатол защищает β-клетки от токсического действия про-воспалительных цитокинов. Перевиваемые культуры β-клеток βTC6 (А, Б, Д) и RIN-5AH (В, Г) инкубировали с брусатолом (50 нМ) совместно с IL-1β и IFNγ в течение 24 ч. Клетки метили иодистым пропидием (PI) (Б, В) или DCFDA (2’-7’-дихлородигидрофлуоресцеин диацетат) (Г). Уровень поглощения клетками PI, что выявляет некротические клетки, и окисления DCFDA, что выявляет клеточный уровень АФК, определяли с помощью проточной цитометрии. Приведены гистограммы уровней живых и пораженных клеток (Б, В) и выработки активных форм кислорода АФК (Г). На панели Д показана экспрессия белка iNOS, определявшаяся методом блот-гибридизации со специфическими антителами. Приведены структура брусатола и изображение его природного источника, тропического кустарника Brucea javanica.
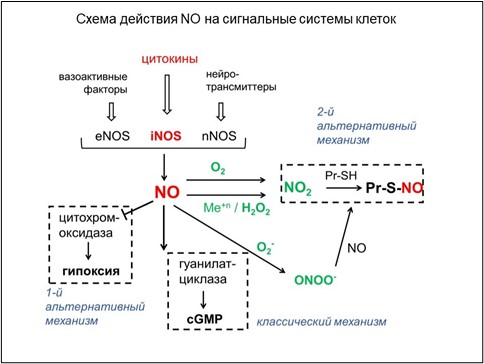
Рисунок 4. Схема классического (cGMP-зависимого) и альтернативных путей сигнального действия NO. Образование NO в клетках происходит при окислении аргинина кислородом в реакции, катализируемой изоферментами NO-синтазы (eNOS, nNOS, iNOS). Действие NO на клетки-мишени опосредовано рядом сигнальных систем. Классический путь активации зависимых от NO сигнальных процессов основан на стимуляции растворимой гуанилатциклазы. Основные альтернативные пути обусловлены (1) подавлением клеточного дыхания в результате связывания NO с гемовой группой цитохромоксидазы, что отчасти воспроизводит состояние гипоксии (т.н. химическая гипоксия) и (2) модификацией SH-групп белков (Pr-SH) в реакции тиольных групп с окисленными производными NO, также обозначаемыми как активные формы азота (АФА). Образование АФА происходит при окислении NO молекулярным кислородом, супероксидом, другими активными формами кислорода (АФК) и ионами металлов.
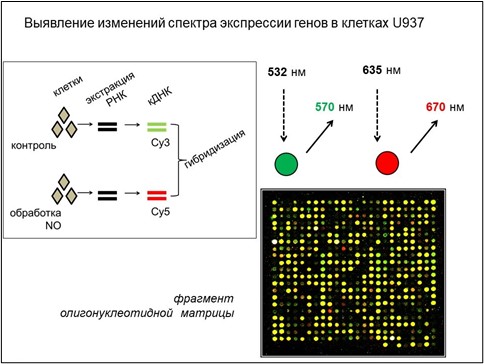
Рисунок 5. Выявление зависимых от NO генов в культуре моноцитов U937. Схема олигонуклеотидной матрицы (cDNA microarray slide). Внизу справа с увеличением показан один из 48 секторов олигонуклеотидной матрицы, которая содержит фрагменты ДНК 26 тысяч генов человека. Каждая точка соответствует флуоресцентному сигналу, основанному на высоко избирательном связывании ДНК матрицы с флуоресцентно меченой кДНК того же гена. Красная, зеленая и желтая окраски указывают на гены, экспрессия которых после инкубации клеток с генерирующими NO соединениями, соответственно, увеличена, снижена или не изменилась. На левой верхней панели показана схема эксперимента. Флуоресцентные метки вносилась в кДНК при синтезе на основе тотальной мРНК, выделенной из контрольных клеток (мечение Cy3) или клеток, инкубированных с донором NO (мечение Cy5). Изменение соотношения флуоресценции Cy3/Cy5 указывает на изменении уровня мРНК соответствующих генов после инкубации клеток с донором NO.
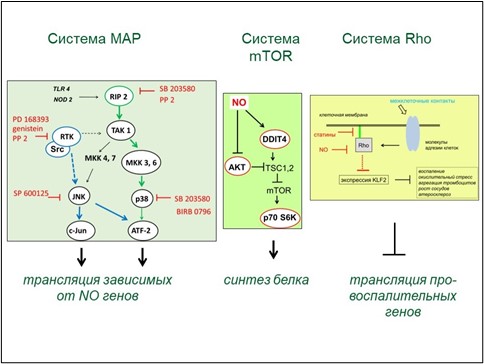
Рисунок 6. Чувствительные к NO сигнальные системы в моноцитах человека. Левая панель. Согласно общепринятым представлениям, активация протеинкиназы RIP2 происходит при связывании мембранных рецепторов TLR4 и NOD2 с лигандами бактериального происхождения (LPS, MDP). Далее RIP2 активирует каскады MAP киназ и модулирует активность фактор NF-κB (не показано). В нашей работе мы выявили активацию RIP2 под действием доноров NO, независимую от лигандов и определяемую по аутофосфорилированию остатка Ser176. В зависимый от RIP2 сигнальный каскад входят контролирующие p38 протеинкиназы MKK3/6 и другие киназы MAP киназ (MKK4/7, TAK1). Фосфорилирование RIP2 подавляют ингибиторы SB 203580 и PP2. Средняя панель. Зависимый от NO цитостатический фактор DDIT4 (DNA damage induced transcript 4) активирует фактор TSC1,2, который действует как негативный регулятор контролирующей синтез белка протеинкиназы mTOR. Протеинкиназа AKT, которую, по нашим данным, ингибирует NO, в свою очередь ингибирует TSC1,2 и восстанавливает активность mTOR. Тем самым, итоговый ответ клеток на действие NO состоит в подавлении синтеза белка. На рисунке обведены элементы регуляторных систем, зависимость которых от доноров NO была нами исследована. Красным цветом выделены ингибиторы, подавляющие зависимую от NO активацию регуляторных протеинкиназ. Правая панель. Доноры NO активируют экспрессию транскрипционного фактора KLF2. Возможно, индукция KLF2 обусловлена подавлением G-белка Rho под действием NO. В присутствии статин GGTI-298 происходит значительное дополнительное усиление NO-зависимой индукции гена KLF2. Известно, что GGTI-298 ингибирует прикрепление G-белков семейства Rho к плазматической мембране, что исключает их участие в сигнальных процессах.
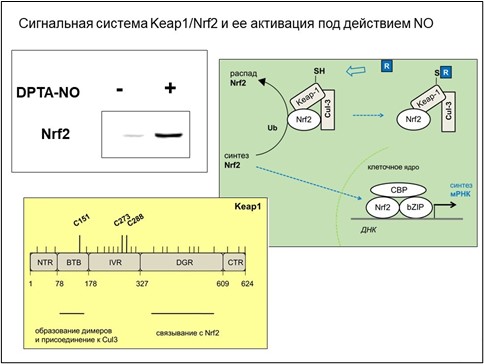
Рисунок 7. Регуляция экспрессии генов, зависимых от системы Keap1/Nrf2. В отсутствие электрофильных соединений убиквитинлигаза E3 (представляет собой комплекс многофункционального белка Keap1, каркасного белка Cul-3 и, собственно, убиквитинлигазы Rbx1) поддерживает клеточное содержание транскрипционного фактора Nrf2 на низком уровне. Под действием окисленных производных NO и электрофилов происходит инактивация Keap1 и подавление убиквитинлигазы, что делает возможным перемещение новообразованного Nrf2 в клеточное ядро и последующую индукцию зависимых генов (например, HO-1 и NQO1). Регуляторные области этих генов содержат участки связывания Nrf2 (регуляторная последовательность ARE – antioxidant response element). Nrf2 связывается с ДНК в составе парного комплекса с одним из транскрипционных факторов семейств Maf или Jun - правая верхняя панель. Инактивация Keap1 происходит при модификации электрофильными соединениями (на рисунке - R) высоко реактивных рецепторных тиольных групп (остатки цистеина C151, C273, C288; не рецепторные остатки цистеина обозначены штрихами) – левая нижняя панель. Пример активации Nrf2 при инкубации клеток U937 с генерирующим NO соединением DPTA-NO (0,2 мМ) - левая верхняя панель
1. Elksnis A., Schiffer T.A., Palm F., Wang Y., Cen J., Turpaev K., Ngamjariyawat A., Younis S., Huang S., Shen Y., Leng Y., Bergsten P., Karlsborn T., Welsh N., Wang X. Imatinib protects against human beta-cell death via inhibition of mitochondrial respiration and activation of AMPK. Clin Sci (Lond). 2021. 135:2243-2263.
2. К.Т Турпаев. Транскрипционный фактор KLF2 и его роль в регуляции воспалительныъ процессов (обзор). Биохимия. 2020. 85: 54-67.
3. Turpaev K, Krizhanovskii C, Wang X, Sargsyan E, Bergsten P, Welsh N. The protein synthesis inhibitor brusatol normalizes high-fat diet-induced glucose intolerance in male C57BL/6 mice: role of translation factor eIF5A hypusination. FASEB J. 2019. 33: 3510-3522.
4. К.Т. Турпаев. Translation factor modification with hypusine and role in regulation of gene expression. as a target for pharmacological interventions Фактор трансляции eIF5A, модификация гипузином и регуляция экспрессии генов. eIF5A как мишень для фармакологических воздействий (обзор). Биохимия. 2018. 83: 863-873.
5. Turpaev K., Welsh N. Aromatic malononitriles stimulate the resistance of insulin-producing βTC6 cells to oxidants and inflammatory cytokines. Eur. J. Pharmacol. 2016. 784: 69-80.
6. Turpaev K., Welsh N. Brusatol inhibits the response of cultured beta-cells to pro-inflammatory cytokines in vitro. Biochem. Biophys. Res. Comm. 2015. 460: 868-872.
7. Ngamjariyawat A., Turpaev K., Vasylovska S., Kozlova E.N., Welsh N. Co-culture of neural crest stem cells (NCSC) and insulin producing beta-TC6 cells results in cadherin junctions and protection against cytokine-induced beta-cell death. PloS One. 2013. 8: 1828.
8. Mokhtari D., Al-Amin A., Turpaev K., Li T., Idevall-Hagren O., Li J., Wuttke A., Fred R.G., Ravassard P., Scharfmann R., Tengholm A., Welsh N. Imatinib mesilate-induced phosphatidylinositol 3-kinase signalling and improved survival in insulin-producing cells: role of Src homology 2-containing inositol 5'-phosphatase interaction with c-Abl. Diabetologia. 2013. 56: 1327-1338.
9. К.Т. Турпаев. Keap1-Nrf2 signaling pathway: mechanisms of regulation and role in protection of cells against toxicity caused by xenobiotics and electrophiles Сигнальная система Keap1-Nrf2, механизм регуляции и роль в защите клеток от токсического действия ксенобиотиков и электрофилов (обзор). Биохимия). 2013. 78:111-126.
10. K. Turpaev, M. Ermolenko, T. Cresteil, JC. Drapier. Potency of benzylidenemalononitriles as heme oxygenase 1 inducers and protectors against oxidative stress. Structure-activity correlations. Biochem. Pharmacol., 2011, 82: 535-547.
11. K. Turpaev, A. Glatigny, J. Bignon, H. Delacroix, JC. Drapier. Variation in gene expression profiles of human monocytic U937 cells exposed to various fluxes of nitric oxide. Free Rad. Biol. Med., 2010, 48: 298-305.
12. K. Turpaev, JC. Drapier. Stimulatory effect of benzylidenemalononitrile tyrphostins on expression of NO-dependent genes in U937 monocytic cells. Eur. J. Pharmacol., 2009, 606: 1-8.
13. Е.А.Мануйлова, К.Т. Турпаев, Е.В. Панкратова. Окись азота подавляет транскрипцию гена гистона H2B. Мол. Биология, 2007, 41: 634-639.
14. К.Т. Турпаев. Роль фактора транскрипции AP-1 в интеграции клеточных сигнальных систем (обзор). Мол. Биология, 2006, 40: 945-961.
15. K. Turpaev, C. Bouton, A. Diet, A. Glatigny, JC. Drapier - Analysis of differentially expressed genes in nitric oxide-exposed human monocytic cells. Free Rad. Biol. Med. 2005, 38: 1392-1400.
16. К.Т. Турпаев, В.С. Прасолов, С. Бутон, Ж.К. Драпье, Д.Ю. Литвинов. Влияние антиоксидантов на зависимую от NO индукцию гена гемоксигеназы 1 в моноцитах U937. Мол. Биология, 2005, 39: 89-95.
17. K. Turpaev, D. Litvinov. Extracellular catalase induces cyclooxygenase 2, interleukin 8, and stromelysin genes in primary human chondrocytes. Biohimie, 2004, 86: 945-950.
18. K. Turpaev, C. Bouton, JC. Drapier. Nitric oxide-derived nitrosating species and gene expression in human monocytic cells. Biochemistry, 2004, 43, 10844-10850.
19. К.Т. Турпаев, Д.Ю. Литвинов. Редокс-зависимая регуляция генов индуцируемых окисью азота (обзор). Мол. Биология, 2004, 38: 56-58.
20. K. Turpaev, D. Litvinov, J. Justesen. 2003. Redox modulation of NO-dependent induction of interleukine 8 gene in monocytic U937 cells. Cytokine, 2003, 23: 15-22.
21. Д.Ю. Литвинов, В.И. Дубовая, М.Г. Васильев, М.В. Лекишвили, В.С. Прасолов, К.Т. Турпаев. Влияние каталазы на экспрессию NO-зависимых генов в первичных хондроцитах. Мол. Биология, 2003, 37: 482-485.
22. К.Т. Турпаев. Активные формы кислорода и регуляция экспрессии генов (обзор). Биохимия, 2002, 67: 281-292.
23. А.И. Николаев, В.И. Дубовая, Д.Ю. Литвинов, А.Б. Полтараус, Д.С. Иванов, А.М. Амченкова, А.Н. Наровлянский, А.Ф. Панасюк, В.С. Прасолов, К.Т. Турпаев. Поиск и идентификация генов индуцируемых в хондроцитах под действием окиси азота. Мол. Биология, 2002, 36: 833-841.
24. K. Turpaev, D. Litvinov, V. Dubovaya, A. Panasyuk, D. Ivanov, V. Prassolov. Induction of vascular endothelial growth factor by nitric oxide in cultured human articular chondrocytes. Biochimie, 2001, 83: 515-522.
25. K. Turpaev, R. Hartmann, J. Justesen. 2`-adenylated derivatives of Ap3A activate RNase L. FEBS Letters, 1999, 457: 9-12.
26. И.Л. Иванова, В.И. Попенко, Н.Е. Черни, А.Н. Наровлянский, А.М. Амченкова, К.Т. Турпаев. Внутриклеточная локализация триптофанил-тРНК синтетазы в линии клеток моноцитов человека после обработки интерфероном. Мол. Биология. 1998, 32: 875-882.
27. К.Т. Турпаев. Роль окиси азота в передаче сигнала между клетками (обзор). Мол. Биология. 1998, 32: 581-591.
28. K. Turpaev, R. Hartmann, L. Kisselev, J. Justesen. Ap3A and Ap4A are primers for oligoadenylate synthesis catalyzed by interferon-inducible 2-5A synthetase. FEBS Letters, 1997, 408: 177-181.
29. K.T. Turpaev, A.M. Amchenkova, A.N. Narovlyansky. Two pathways of the nitric oxide-induced cytotoxycal action. Biochem. Molec. Biol. Intern. 1997, 41: 1025-1033.
30. K.T. Turpaev, V.M. Zachariev, I.V. Sokolova, A.N. Narovlyansky, A.M. Amchenkova, J. Justesen, L.Yu. Frolova. Alternative processing of the tryptophanyl-tRNA synthetase mRNA from interferon-treated human cells. Eur. J. Biochem., 1996, 240: 732-737.
31. A.A. Vartanian, A.N. Narovlyansky, A.M. Amchenkova, K.T. Turpaev, L.L.Kisselev Interferons induce accumulation of diadenosine triphosphate (Ap3A) in human cultured cells. FEBS Letters. 1996, 381: 32-34.
32. К.Т. Турпаев, В.М. Блинов, Л.Л. Киселев. Короткая 5’-концевая последовательность про-мРНК триптофанил-тРНК синтетазы подавляет сплайсинг альтернативных экзонов. Доклады РАН 1996, 349: 698-700.
33. И.В. Соколова, А.Н. Наровлянский, А.М. Амченкова, К.Т. Турпаев. Альтернативный сплайсинг 5’-концевых экзонов гена триптофанил-тРНК синтетазы человека. Мол. Биология. 1992, 30: 319-329.
34. А.А. Вартанян, К.Т. Турпаев, А.Н. Наровлянский, А.М. Амченкова, Л.Л. Киселев. Интерфероны индуцируют накопление диаденозинтрифосфата (Ap3A) в моноцитах человека. Доклады РАН 1995, 344: 252-255.
35. К.Т. Турпаев, А.В. Горюнов. Статистический анализ энхансеров ряда эукариотических генов. Мол. Биология. 1992, 26: 462-474.
36. K.T. Turpaev, A.V. Itkes, N.M. Alexandrova, O.V. Pokrovskaya, L.R.Imamova, B.K. Chernov, and L.L. Kisselev - Binding of proteins of HeLa cell extract to oligonucleotides containing consensus IRS and to IRS-containing fragment of human c-myc gene. - Eur. J. Biochem., 1991, 200: 107-111.
37. Y.S. Vassetzky, K.T. Turpaev, M.A. Lagarkova, S.V. Razin. Characterization of a novel chicken repeat that contains both a nuclear matrix attachment site and a recognition sequence for a novel soluble DNA-binding protein. The Nucleus, 1991, 34: 49-52.
38. К.Т. Турпаев, Е.С. Васецкий. Ядерные белковые факторы, узнающие специфические последовательности ДНК. Обзор. Генетика 1990, 804-816.
39. К.Т. Турпаев, В.В. Адлер, С.В. Разин. Специфическое связывание растворимых ядерных белков с клонированными фрагментами постоянно прикрепленной к ядерному скелету ДНК. Доклады АН СССР 1990, 314: 1260-1263.
40. А.Н. Наровлянский, К.Т. Турпаев, А.М. Амченкова, А.В. Иткес, Е.Г. Алимов. Индукция интерферон-специфических ферментов в чувствительных и устойчивых к интерферону клетках. Мол. Генетика. Микробиол. Вирусол. 1988 (2): 34-39.
41. А.И. Бохонько, К.Т. Турпаев, А.В. Иткес, Т.В. Мамонтова, Т.Г. Орлова. Эффект модуляции продукции интерферона в обработанных интерфероном клетках L-929. Мол. Генетика. Микробиол. Вирусол. 1988 (2): 28-33.
42. К.Т. Турпаев, А.В. Тимофеев, К.А. Кафиани – Активация 2-5 олиго(А) синтетазы при тепловом воздействии на культуру фибробластов хомячка. Доклады АН СССР 1987, 297: 737-740.
43. A.V. Itkes, O.N. Kartasheva, V.L. Tunitskaya, K.T. Turpaev, C.A. Kafiani, and E.S. Severin - Activities of cAMP- dependent protein kinase and enzymes of 2`,5`-oligoadenylate metabolism in NIH 3T3 cells deepening into the resting state. - Exp. Cell Res., 1985, 157: 335-342.
44. E. S. Severin, A.V. Itkes, O.N. Kartasheva, V.L. Tunitskaya, K.T. Turpaev, and C.A. Kafiani - Regulation of 2-5 A phosphodiesterase activity by cAMP- dependent phosphorylation: mechanism and biological role. - Adv. Enz. Reg., 1985, 23: 365-376.
45. О.Н. Карташева, А.В. Иткес, К.Т. Турпаев, В.Л. Туницкая, Е.С. Северин. Исследование 2’-фосфодиэстеразной активности в культуре NIH 3T3 при активации цАМФ-зависимого фосфорилирования. Мол. Биология. 1985, 19: 450-455.
46. A.V. Itkes, K.T. Turpaev, O.N. Kartasheva, V.L. Tunitskaya, and C.A. Kafiani - The mechanisms of the cyclic AMP- dependent regulation of the enzymes of the 2`,5`-oligoadenylate system. - FEBS Lett., 1984, 171: 101-105.
47. К.Т. Турпаев, А.В. Иткес, О.Н. Карташева, В.Л. Туницкая, К.А. Кафиани – Циклический АМФ регуляция 2-5 олиго(А) синтетазной активности в культуре клеток фибробластов мыши. Доклады АН СССР 1984, 275: 1231-1234.
48. A.V. Itkes, K.T. Turpaev, O.N. Kartasheva, C.A. Kafiani, and E.S. Severin. Cyclic AMP-dependent regulation of synthesis and phosphodiesterase of 2`,5`- oligoadenylate in NIH 3T3 cells. Mol. Cell. Biochem., 1984, 8: 165-171.
49. A.V. Itkes, K.T. Turpaev, O.N. Kartasheva, V.L. Tunitskaya, C.A. Kafiani, E.S. Severin. Regulation of 2`,5`-oligo(A) synthetase activity in theophylline-treated NIH 3T3 cells. FEBS Lett., 1984, 166: 199-201.
50. C.A. Kafiani, A.V. Itkes, O.N. Kartasheva, M.N. Kochetkova, K.T. Turpaev, E.S. Severin. A study of the relationship between the interferon enzyme system and the system of the cyclic nucleotide metabolism. - Adv. Enz. Reg., 1983, 21: 353-365.
51. A.V. Itkes, K.T. Turpaev, O.N. Kartasheva, A.I. Vagonis, C.A.Kafiani, E.S. Severin. Theopylline-induced 2`,5`-oligo(A) synthetase in NIH 3T3 cells. Biochem. Intern., 1982, 5: 15-21.